
On May 6, 2024, the U.S. Food and Drug Administration (FDA) issued a final rule that significantly modified its approach to overseeing laboratory developed tests (LDTs). This rule, which became effective on July 5, 2024, marks a pivotal change for companies producing LDTs. The new regulations aim to ensure that these tests meet the same standards for safety and reliability as other invitro diagnostic tests.
While this increased oversight will enhance patient safety and test reliability, these regulations will require changes to existing lab processes and workflow, roles and ways of working, and lab systems. Laboratories must begin taking action now to align with the new regulations and minimize potential disruptions.
What Are LDTs, and why is the FDA making regulatory changes to them?
Laboratory-developed tests (LDTs) are diagnostic tests designed, manufactured, and used within a single laboratory to measure or detect substances such as proteins, glucose, cholesterol, or DNA in human specimens. Historically, the FDA has exercised enforcement discretion over LDTs, considering them low-risk due to their limited use and simpler technology.
However, the landscape has changed dramatically. Many LDTs now employ advanced technology, are used in high volumes, and are marketed nationwide. This evolution has prompted the FDA to enforce stricter regulations to ensure these complex tests meet appropriate safety and efficacy standards. While LDTs previously fell into a gray zone, they are now classified as medical devices and are subject to premarket review requirements, quality system regulations, medical device reporting, and other regulatory controls.
FDA’s “The Final Rule” and the Key Change to LDT regulations
The final rule amends FDA regulations to explicitly classify in vitro diagnostic products (IVDs), including LDTs, as devices under the Federal Food, Drug, and Cosmetic Act (FD&C Act). This change initiates a phased implementation of IVD requirements over four years, gradually increasing oversight and compliance expectations for LDTs.
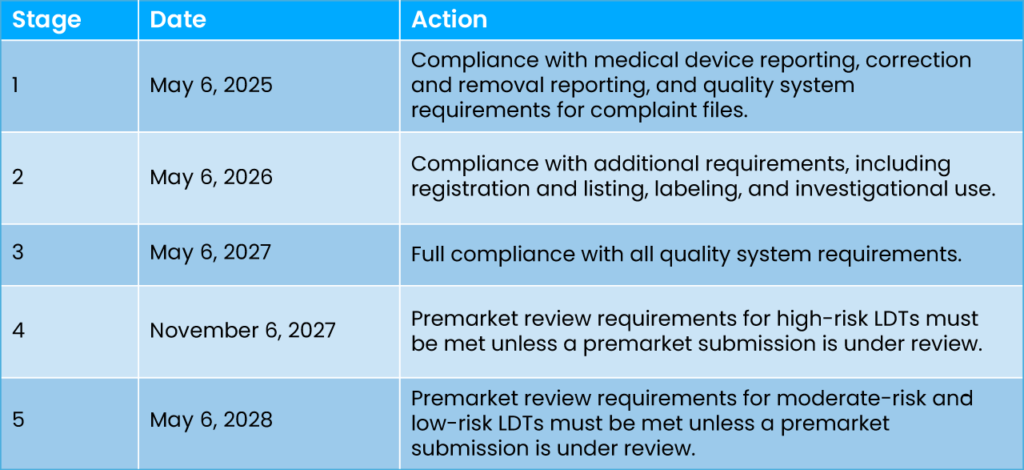
Phased Implementation Highlights
LDT manufacturers will now be required to comply with a wide array of medical device regulatory requirements, including premarket review, quality system regulations, and adverse event reporting. The phased implementation is designed to help laboratories transition smoothly to the new regulatory framework. Here’s a breakdown of the stages:
Impact on Medical Device Companies
The new rule will have significant implications for companies producing medical devices for LDTs:
- Operational Changes: Companies will need to adjust their operations to meet new compliance requirements, including enhanced documentation and reporting processes. These operational changes will likely drive higher cost per test.
- Regulatory Strategy: Engaging with the FDA early and planning for phased compliance will be crucial. Companies should consider pre-submission meetings and other forms of FDA engagement to clarify regulatory pathways.
- Quality Systems: Enhancements to quality management systems will be necessary to meet FDA standards, particularly in areas such as design controls, purchasing controls, and corrective actions.
- Premarket Submissions: Companies must prepare for the premarket review process, especially for moderate- to high-risk LDTs, to ensure timely compliance.
- Market Dynamics: Increased regulatory oversight may shift market dynamics, potentially affecting competition and innovation within the industry.
Preparing for Compliance
Compliance with the new regulations is a necessity, not a choice. To navigate these changes successfully, laboratories must take proactive steps:
- Early Engagement: Engage with the FDA early to understand specific requirements and timelines.
- Resource Utilization: Use all available FDA resources, including webinars, guidance documents, and templates, to aid in compliance efforts.
- Strategic Planning: Develop a comprehensive compliance strategy that addresses each stage of the phased implementation, ensuring all regulatory requirements are met on time.
- Program and Change Management: Appoint an implementation lead to build and monitor a comprehensive delivery and change plan to prepare the organization and its personnel for this transformation.
Conclusion
The FDA’s new rule on LDTs represents a significant shift in regulatory oversight, aimed at enhancing the safety and reliability of these tests. For labs producing LDTs, proactive planning and change management are essential to ensure a smooth transition and continued market presence. By engaging with the FDA, leveraging available resources, and establishing a focused program, labs can navigate these changes effectively and maintain their competitive edge in the evolving landscape of diagnostic testing.

Amy Flynn is a Managing Director with alliantConsulting. With over three decades of experience in the pharmaceutical, medical device, and diagnostic industries, Amy’s expertise spans various business functions, from clinical and regulatory, to marketing and business development. Her career includes roles as Global and National Life Sciences Industry Lead at Grant Thornton and General Manager of Genomics at Whatman Biosciences, as well as founding partner of CatMa Consulting. She has led major change initiatives, mergers and acquisitions, and quality systems implementations. Amy has an M.Ed. in Counseling Psychology from Temple University, as well as an M.B.A. and a B.S. in Engineering from Rutgers College of Engineering. She also holds certifications in change management and leadership coaching, and has been recognized as an HBA Life Science Luminary and a Consulting Report Top 50 Consultant.








